 RSS Feed
RSS Feed

















 September 1st, 2022
September 1st, 2022  Awake Goy
Awake Goy Hundreds of thousands of peer-reviewed papers in genetics employ a method called Principal Component Analysis. But new research shows this method is highly biased. This means that multitudes of major studies regarding ancient populations may be drastically wrong!
The Irrisistable Draw of PCA
It’s hard to make new friends, especially once you are past your thirties. As Seinfeld so eloquently put it: Whatever the group is that you’ve got now, that’s who you’re going with. You’re not interviewing, you’re not looking at any new people, you’re not interested in seeing any applications . For DNA scientists, and scientists as a whole, the situation is even worse. The long hours and isolation required to do our research have determinetal effects on our social lives. Of course, there are always exceptions. Sometimes you get to know someone who is always there to support you, someone who asks for little and gives much. Someone you can always show up to a party with and be proud of. Someone that your friends and collaborators will admire because it makes you look smart and cool with deep understanding of the science involved. Who doesn’t want a friend like that? Replace “someone” with “something” and you will understand what Principal Component Analysis (PCA) is to scientists, specifically, to population geneticists.
What is Principal Component Analysis?
PCA is a mathematical transformation that takes a complex dataset, like 10,000 genomes of 2,000 people around the world, and transforms it so that it can be represented by a colorful X-Y scatter plot at the click of a button. It is the best friend of the procrastinating student who has a conference tomorrow and needs to slap some results together quickly, the lecturer looking to produce papers in a rush, and the professor looking for a promotion by making hyped claims without evidence. The number of friends that PCA has is a reminder of the good ol’ MySpace days – with citations numbering around 200,000 in genetics alone, times an average number of 10 authors per paper, we get 2,000,000 scholars who authored a paper that used PCA.
PCA is used to examine the population structure of a group of individuals to determine their ancestry, analyze the demographic history and admixture, decide on the genetic similarity of individuals and exclude outliers, decide how to model populations, describe the ancient and modern genetic relationships between individuals, infer familial relatedness, identify ancestral trends in the data, detect genomic signatures of natural selection , identify evolutionary trends , support genetic studies of diseases, geolocalize individuals, draw historical and ethnobiological conclusions, and more. It is “The Little Scatterplot that Could.”
The problem with PCA was also its greatest advantage. It always told everyone what they wanted to hear, so no one dared to challenge it. So, naturally, I did.
PCA: A Dubious Method?
In a paper published in Scientific Reports , I showed that PCA results are far more sensitive to the input than anyone has appreciated. By way of analogy, think of PCA as an oven with flour, sugar, and eggs as the data input. The oven may always do the same thing, but the outcome, a cake, critically depends on the ratio of ingredients and how they are combined. In just the same way, minor changes to the way data are input causes PCA to generate radically different outputs, leading to incorrect results, misconceptions, and a lack of replication.
One of the fields considered to be best-friends-forever with PCA is paleogenomics, where we want to learn about ancient peoples and individuals such as Copper Age Europeans. It is expected that they would be similar to Europeans, and scientists used PCA to show that Copper Age Europeans cluster with Europeans. Why? Because the rationale of using PCA is that it can be used to create a genetic map that positions the unknown population alongside the populations they are most related to. Because PCA only sees the data (without the labels), we assume that it is a neutral and unbiased tool, and that the answer that it gives is correct.
My study showed that small changes in the number of individuals and choice of populations can produce a very large difference in the PCA results, allowing the experimenter full control of the results.
In this way, the experimenter (in this case, me) can produce very different answers to the simple question “Which population are Copper Age Europeans closest to genetically?”, by placing them close to any population. I did that by changing the number of individuals in each population (Oceanians, South Asians, etc.) and choosing different sub-populations. What happened? Our supposedly unbiased tool, the geneticists’ compass, produced four different historical scenarios (out of practically endless historical versions) all mathematically ‘correct’, but only one may be biologically correct (if at all).
Such ‘conclusions’ are derived from PCA in almost any genetic population paper on humans, plants, animals, medical genetics, and drug testing (where cases and controls are matched). PCA results are not limited to scientific papers. They are also embedded in large datasets, used by genetic testing companies, and utilized to support policy decisions. There is not a single reader who is not impacted by PCA, whether they know what it is or just learned about it now. Up to 216,000 peer-reviewed papers in genetics alone have employed PCA for exploring and visualizing similarities and differences between individuals and populations and based their conclusions on these results.
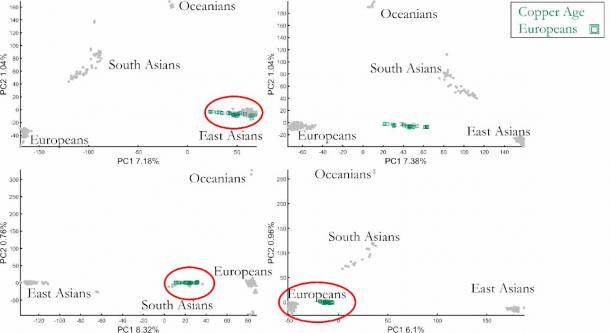
This figure shows four out of endless PCA outcomes describing Copper Age Europeans’ origins. The PCA plots were generated using the same reference populations but with different population sizes allowing anyone to pick their favorite historical scenario. (Author Provided)
Scientific Conclusions May be Drastically Wrong
To put these examples into context, consider the recent publication of “ Ashkenazic Jewish graves from 12th century England ” by Mark G. Thomas (who has been criticized for misappropriating evidnece) and Ian Barnes. This study ‘explores’ the ancestry of six newly-found ancient individuals, and as always, it starts with a PCA plot where the ancient individuals are projected on top of known modern individuals to identify their ancestry (recall that overlap = ancestry).
A few things are immediately obvious from this plot. First, Ashkenazic Jews cluster with Southern Europeans (i.e. they are genetically indistinguishable from them); thus, the entire premise of this paper is flawed. These people could have well been Italians. Second, although three of the ancient individuals are siblings, they do not cluster together, which should already raise concerns about the validity of this approach. Third, there are very few non-Jewish populations at the bottom of the plot, which was done to a) avoid showing that modern-day Jews overlap with modern non-Jews and b) have the ancient individuals overlap with Africans. Finally, there are no other ancient populations that cluster with their respective modern-day populations to convince us that this tool actually works.
We can see that while this plot is presented as an exploration of hypothesis, the experimenters constructed it to yield their desired results, which, annoyingly, it barely did! Nonetheless, the authors concluded that “These results are consistent with the Chapelfield individuals having Jewish ancestry”, citing an irrelevant paper to add credibility to their findings. Despite these problems, and although at no point did these samples overlap with Ashkenazic Jews, they were concluded to be of Ashkenazic descent, and the article was featured in Nature (a for-profit journal) with my short criticism somewhere inside. In this field, truth is as important as the socks you took off your feet yesterday after a long hot day.
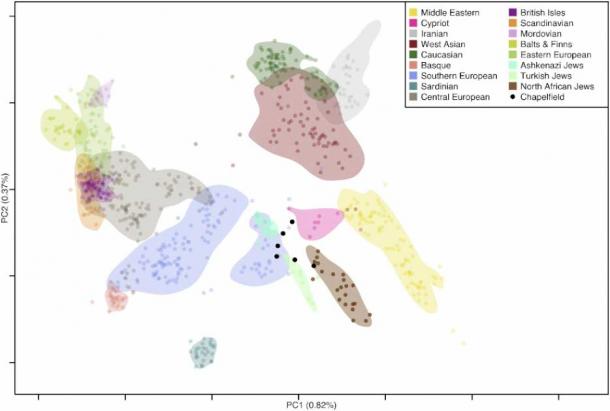
PCA plot of the unknown ancient individuals (black) and known modern-day populations (color) (Author Provided)
PCA is an illustration of dataism in population genetics. Dataism describes an ideology formed by the emergence of Big Data, where measuring the data is the ultimate achievement. Dataism proponents believe that with sufficient data and computing power, the world’s mysteries will reveal themselves. Dataism enthusiasts rarely ask themselves if PCA results are correct, but rather how to interpret the results correctly. As such, clustering is interpreted as identity due to common ancestry, and its absence as genetic drift. In PCA-driven science, almost all the answers are equally acceptable, and the truth is in the eyes of the beholder. While PCA explains nothing, it does illustrate Seinfeld’s point. It’s really hard to make friends when you are old, particularly if you are a scientist.
[embedded content]
Independent comment about the paper
“Techniques that offer such flexibility encourage bad science and are particularly dangerous in a world where there is intense pressure to publish. If a researcher runs PCA several times, the temptation will always be to select the output that makes the best story”, added Professor William Amos, Professor of Evolutionary Genetics at the University of Cambridge, who was not involved in the study.
Top Image: Geneticist contemplating his DNA dataset. Source: Grispb / Adobe Stock

By Eran Elhaik
Related posts:
Views: 0
 Posted in
Posted in  Tags:
Tags: 
















